4 главных метода секвенирования генома

Около 70 лет назад случилось поворотное событие в развитии науки — была открыта молекулярная структура дезоксирибонуклеиновой кислоты. Её двойная спираль не только объяснила механизм хранения наследственной информации, но и открыла путь к изучению молекулярных основ жизни. Узнать, где и какие гены находятся в ДНК, можно, определив последовательность её нуклеотидов. Это называется секвенированием. Разбираемся в том, как оно работает, на примере четырёх основных подходов к расшифровке генома.
«Над этой проблемой бились многие годы. На какие ухищрения только не шли! Предлагали, например, „пришивать“ к каждому нуклеотиду данного сорта, скажем, к адениновому, соединение, содержащее атомы урана, и с помощью электронного микроскопа, в который такие тяжёлые атомы видны, рассмотреть, как эти метки распределены вдоль цепи одиночной нити ДНК. Затем „пришивать“ атомы урана к тиминовым основаниям и т.д. Однако несмотря на многолетние усилия, получить вразумительные результаты не удалось».
Отрывок из книги Максима Франк-Каменецкого «Самая главная молекула. От структуры ДНК к биомедицине XXI века»
 Во время пандемии COVID-19 учёные использовали секвенирование для расшифровки генома вируса, что позволило создать тесты, вакцины и лекарства
Фото: © Александр Щербак / ТАСС
Во время пандемии COVID-19 учёные использовали секвенирование для расшифровки генома вируса, что позволило создать тесты, вакцины и лекарства
Фото: © Александр Щербак / ТАСС
Метод Сенгера
Всё же учёные научились определять последовательности ДНК. На сегодняшний день придумано несколько десятков подходов. Самый популярный из них предложил британский биохимик Фредерик Сенгер. Этот метод основан на копировании небольших кусочков ДНК и добавлении в них специальных «стоп-знаков», которые говорят, когда нужно остановиться. Процесс секвенирования по Сенгеру проходит в несколько этапов.
- К исследуемой цепи ДНК добавляют праймер, нуклеотиды (аденин, тимин, гуанин и цитозин), которые будут присоединяться к нити ДНК и полимеразе.
- Нуклеотиды «включаются» в растущую цепь ДНК, в результате чего получаются фрагменты разной длины.
- Когда нуклеотиды встраиваются в синтезируемую цепь ДНК, дальше она расти не может. По месту её обрыва можно определить последнюю букву в изучаемом фрагменте ДНК.
Фредерик Сенгер — дважды лауреат Нобелевской премии по химии. Впервые он получил её в 1958 году за разработку метода секвенирования белков, а в 1980 году был удостоен второй такой награды — за секвенирование ДНК. Его методы стали основой для дальнейших исследований в молекулярной биологии и генетике. Сенгер также разработал метод, известный как «метод Сенгера», который используется для определения последовательности нуклеотидов в ДНК.
 Фредерик Сенгер
Фото: Public domain
Фредерик Сенгер
Фото: Public domain
Именно благодаря открытию Сенгера начался проект «Геном человека», целью которого стало определение последовательности всех нуклеотидов в геноме. В итоге расшифровать наследственную информацию человека целиком удалось за 13 лет (это произошло в 2007 году). Интересно, что первая «секвенированная» личность — это Джеймс Уотсон, американский биолог, который вместе с коллегой Фрэнсисом Криком в 1972 году и вывел структуру двойной спирали ДНК.
В текущей версии генома насчитывается 2,85 миллиарда нуклеотидов, разделённых 341 пробелом. Эта версия охватывает примерно 99 % эухроматического генома и содержит примерно одну ошибку на 100 тысяч оснований.
Секвенирование нового поколения (NGS)
NGS-секвенирование появилось как ответ на потребность в быстром и массовом чтении ДНК. В отличие от устаревших методов, оно позволяет считывать миллионы фрагментов одновременно. NGS-подход основан на параллельной амплификации множества коротких участков ДНК и регистрации каждого добавленного нуклеотида с помощью светового сигнала или других меток.
 Illumina — модель для NGS-секвенирования
Фото: © MGI Tech Co.
Illumina — модель для NGS-секвенирования
Фото: © MGI Tech Co.
Особенности NGS-секвенирования:
- нить ДНК делят на фрагменты, к которым присоединяют специальные метки — адаптеры, помогающие ей прикрепиться к ячейке секвенатора;
- в ячейках происходит амплификация и достраивание цепей ДНК с помощью нуклеотидов;
- полученные цепочки ДНК загружают в секвенатор, где компьютер собирает все прочитанные фрагменты в единую последовательность и ищет в ней отклонения (например, изменения в участке гена, которые вызвали болезнь).
Нанопоровое секвенирование
Нанопоровое секвенирование использует для расшифровки генома крошечные отверстия — нанопоры. Молекула ДНК, которая имеет отрицательный заряд, при воздействии электрического поля проходит через белковую пору, расположенную в мембране. Это приводит к снижению силы тока из-за уменьшения диаметра отверстия и позволяет определить, какой нуклеотид проходит через нанопору в определённый момент времени.
В далёком будущем экспресс-секвенирование наверняка будет использоваться для открытия сейфа по ДНК владельца, как это бывает в фантастических фильмах
Даниил Задорожный, младший научный сотрудник Научного центра генетики и наук о жизни Университета «Сириус»
Пиросеквенирование
Пиросеквенирование — это метод определения последовательности ДНК с помощью химических реакций, которые сопровождаются вспышками света. Такой подход позволяет определить последовательность нуклеотидов, регистрируя высвобождающиеся при синтезе ДНК пирофосфаты. Так можно «прочитать» фрагменты генома быстро и относительно дёшево, что делает этот метод подходящим для массового секвенирования.
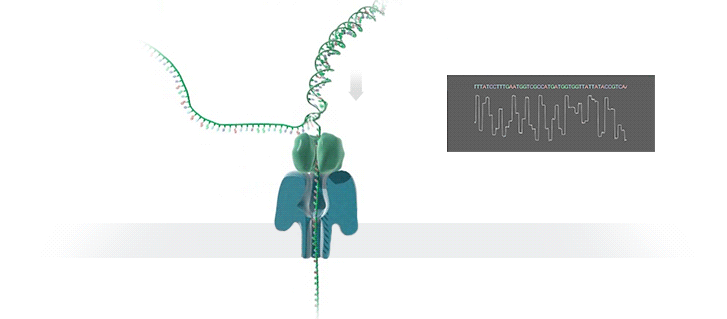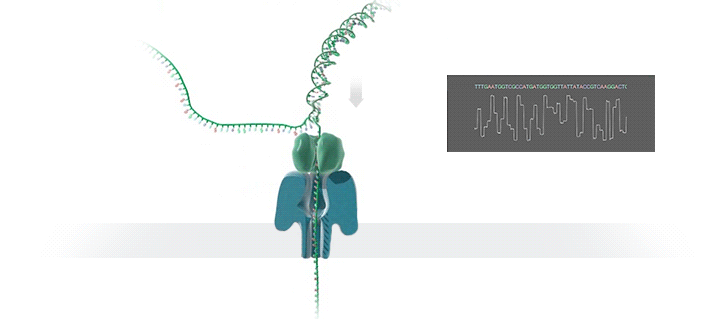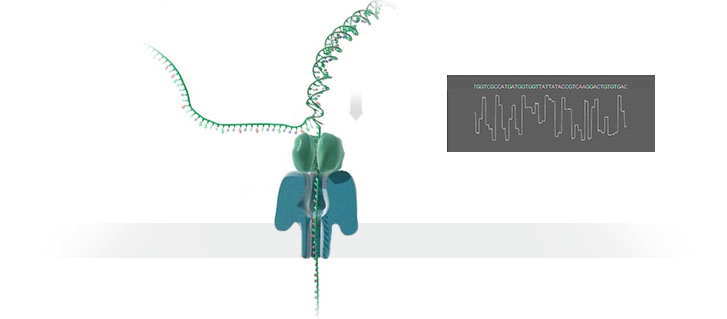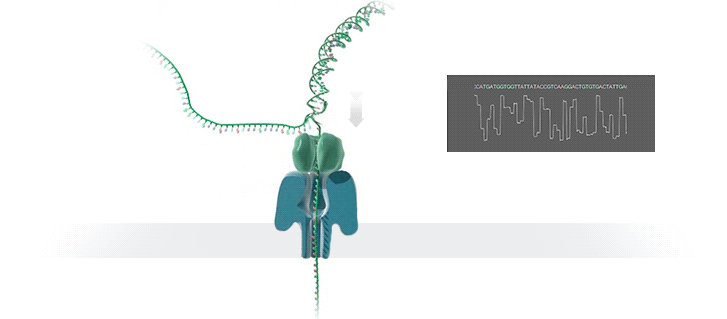 Каждый нуклеотид имеет свою уникальную форму, которая влияет на силу тока
Изображение: © HABR
Каждый нуклеотид имеет свою уникальную форму, которая влияет на силу тока
Изображение: © HABR
«Секвенирование используется не только для прямого определения последовательности, но и для подготовки к другим лабораторным исследованиям.
Например, работа с системой CRISPR/Cas9 также требует предварительного секвенирования — сначала учёные определяют участок ДНК, который хотят изменить, а затем программируют систему CRISPR так, чтобы она точно нашла нужное место в геноме. После редактирования снова проводят секвенирование, чтобы подтвердить успешное изменение и исключить побочные эффекты. Сейчас расшифровку генома пытаются сделать ещё более быстрой и дешёвой, при этом точной. В будущем возможно секвенирование одной клетки в реальном времени, а также использование искусственного интеллекта для анализа геномных данных. Также развиваются методы третьего поколения, которые позволяют читать очень длинные последовательности без сложной подготовки», — говорит Даниил Задорожный.

Главные методы редактирования генома
Читать



